单层InSe能带结构的模拟
操作系统:debian 9
vasp版本:5.4.4
p4vasp:0.3.29r1
VESTA:3.4.4
以下程序不保证在你的计算机上就一定能运行成功,直接复制可能会报错,以下程序主要参考这里。最后附有相关代码。
因为InSe和石墨烯具有相识的结构,所以我们可以先模拟出石墨烯的结构和能带图
首先,在同一个文件夹下准备以下四个文件,INCAR、POSCAR、KPOINTS以及POTCAR
其中POTCAR文件我们可以从赝势库中得到,INCAR文件决定作业做什么和怎么做,里面的详细参数可以参考最下面的附件。
INCAR文件内容如下
SYSTEM = Graphene molecule in a box #注释,必须有,!和#开头都可以
ISTART = 0 #随机初始化波函数
ICHARGE = 2 #ISTART为0时,默认为2,否则为0
ENCUT = 300 #截断能,从C的赝势库中查询,ENMAX=400,ENMIN=300ev
EDIFF = 1E-6
ISMAR = 0
SIGMA = 0.01KPOINTS文件内容如下
K-POINTS #注释,必须有
0 #k点数目,0代表自动生成
G #k点类型,第一个字母有用,表示原点在伽马点的Monkhorst-pach网格,在倒空间中
10 10 10 #网格尺寸
0 0 0 #k点相对于网格原点的平移,倒空间中,一般取为0POSCAR文件包含了晶胞基失和原子坐标
六边形碳,对角线长2.45988埃,如过定义OA=1(实际长1.22994埃),以O原点,OA为x轴建立直角坐标系,则相关坐标能写出来,然后扩展到三维,石墨烯原子层间距为0.97nm(不确定),也就是9.7埃。
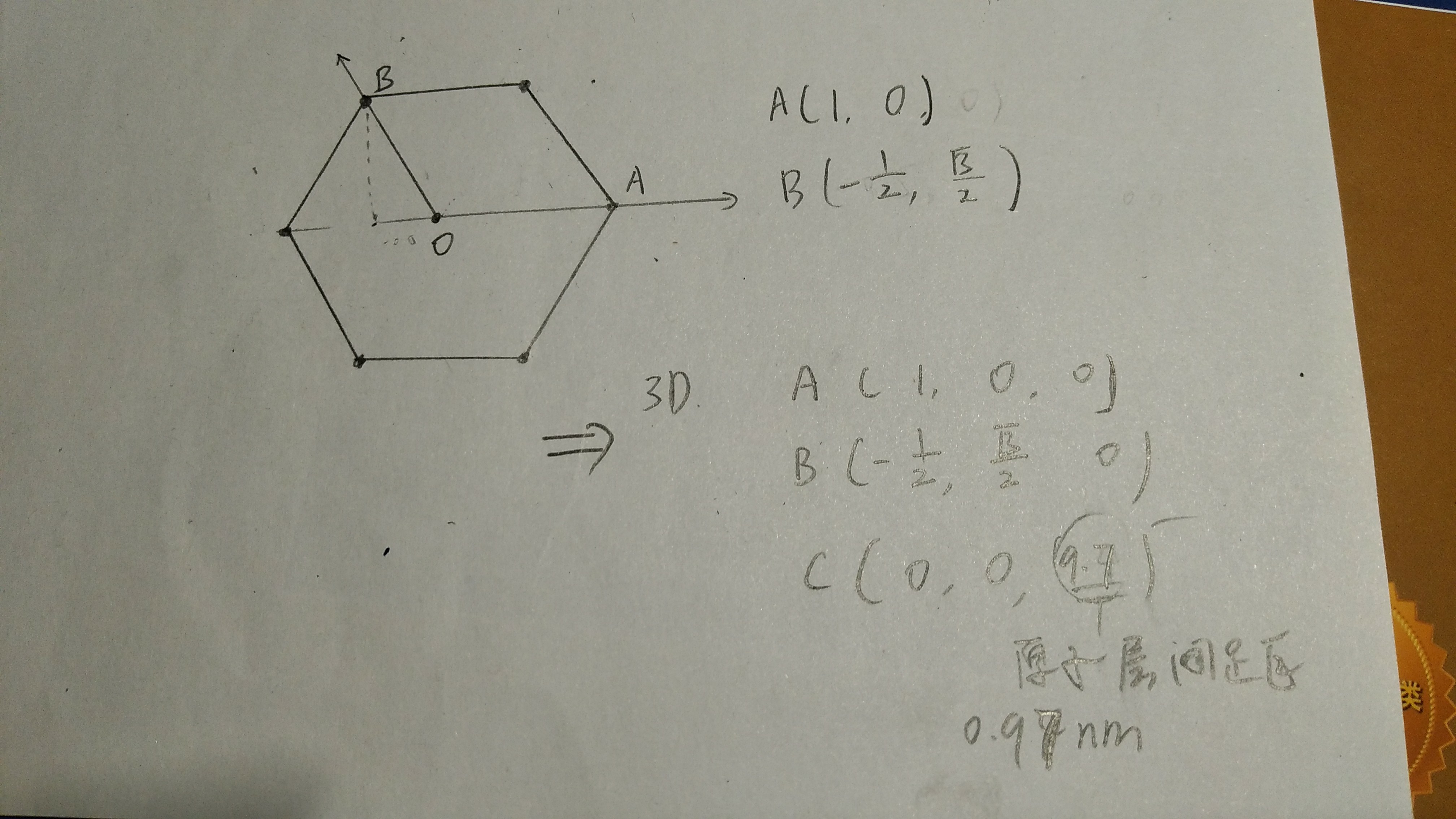
POSCAR文件内容如下
fcc: #注释行
2.45988 #缩放系数,对下面数字都有效,可取为晶格常数。
1.000000 0.000000 0.000000 #晶胞的基失,A,B,C在坐标轴上的投影
-0.50000 0.866025 0.000000
0.000000 0.000000 9.700000
2 #各类原子的数目,相同可以合并,不是同一类,或写成1 1,将两个碳的赝势文件合并,即在赝势文件中重新复制一份到后面
Direct #坐标类型,第一个字母有效,D表示以晶胞基失为单位,在基失下的投影,C代表在直角坐标下的投影
0.000000 0.000000 0.000000 #第一个原子坐标,可任意取
0.333333 0.666667 0.000000 #第二个原子坐标(相对,疑问,如何确定)p4vasp得到的结构
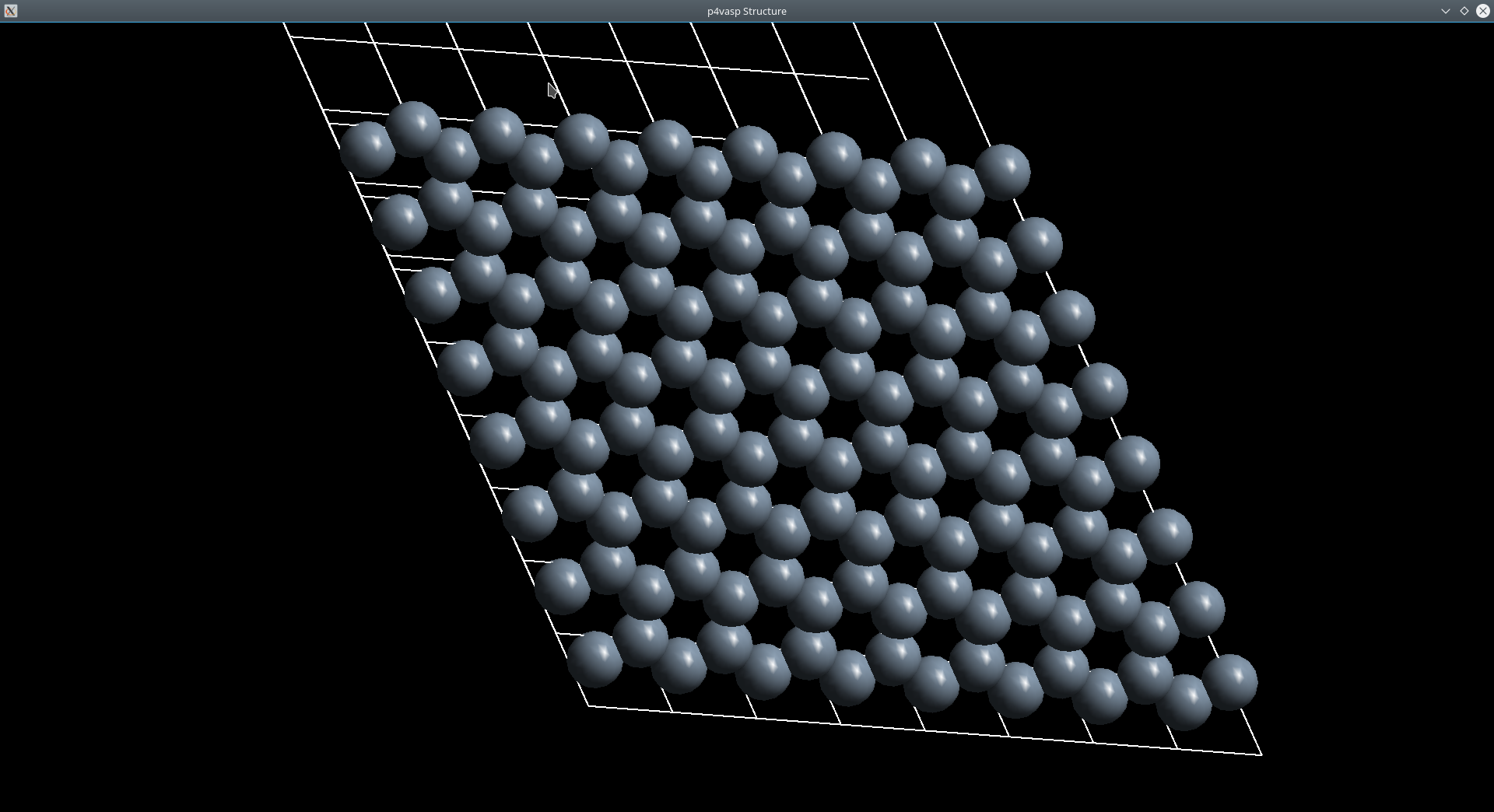
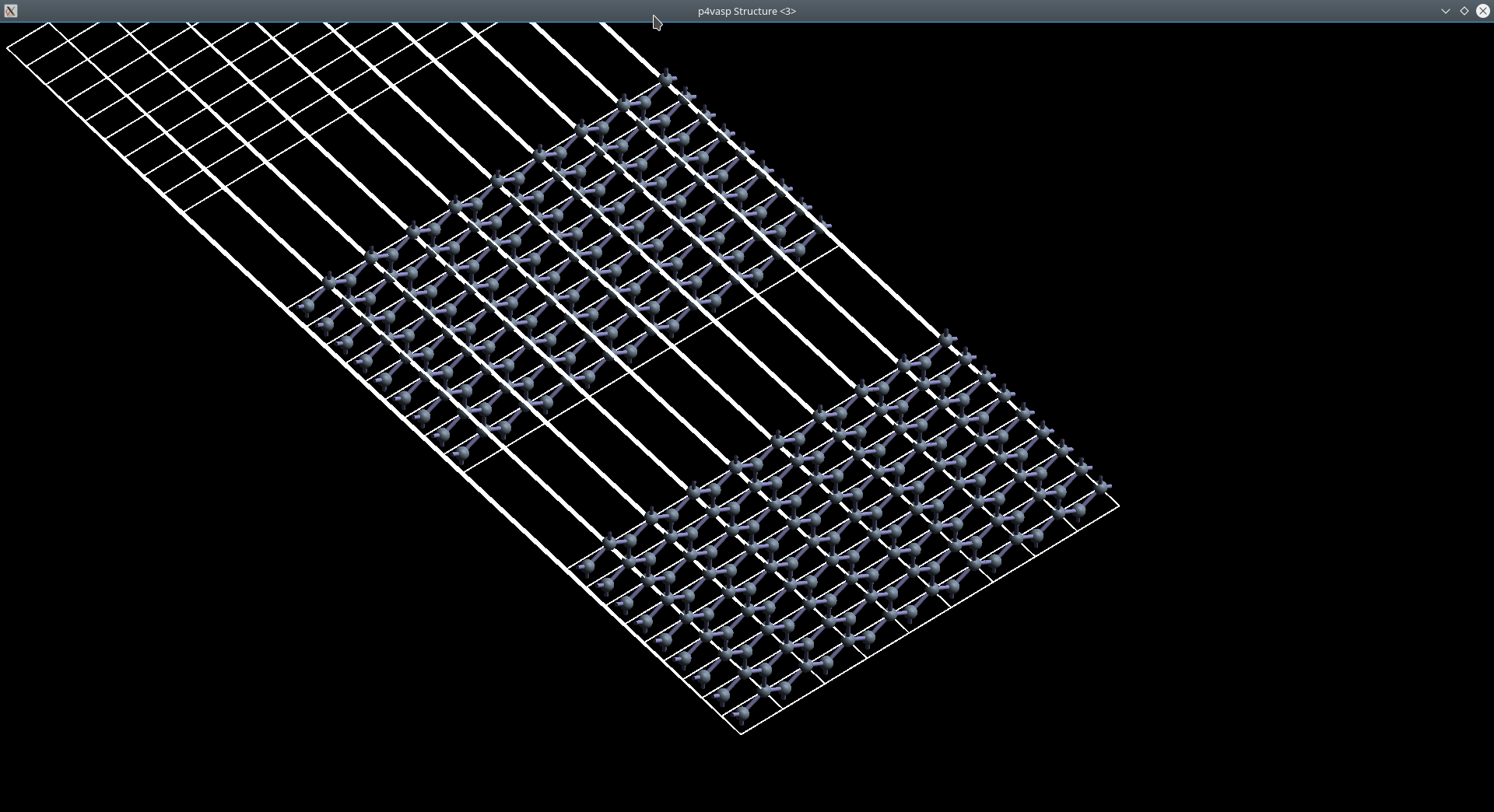
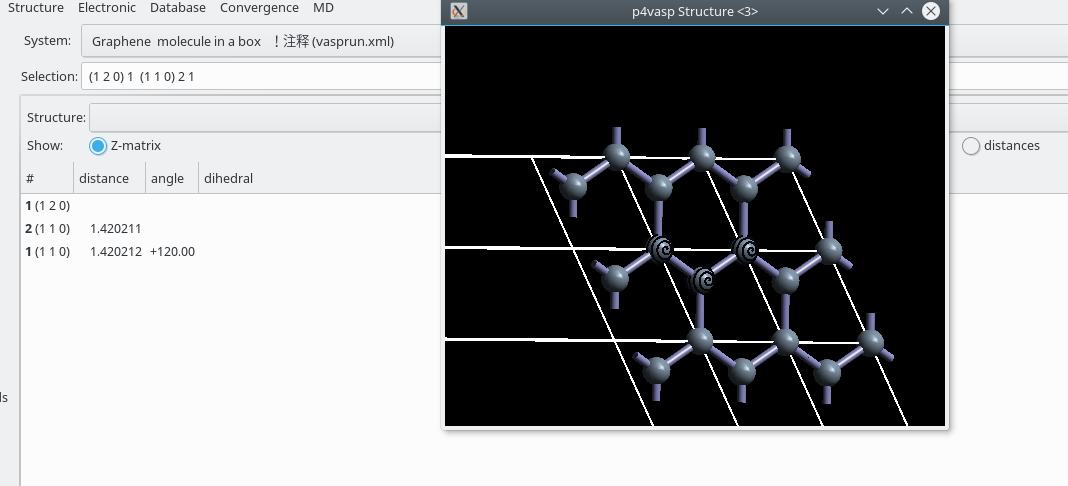
然后是VESTA中得到的图
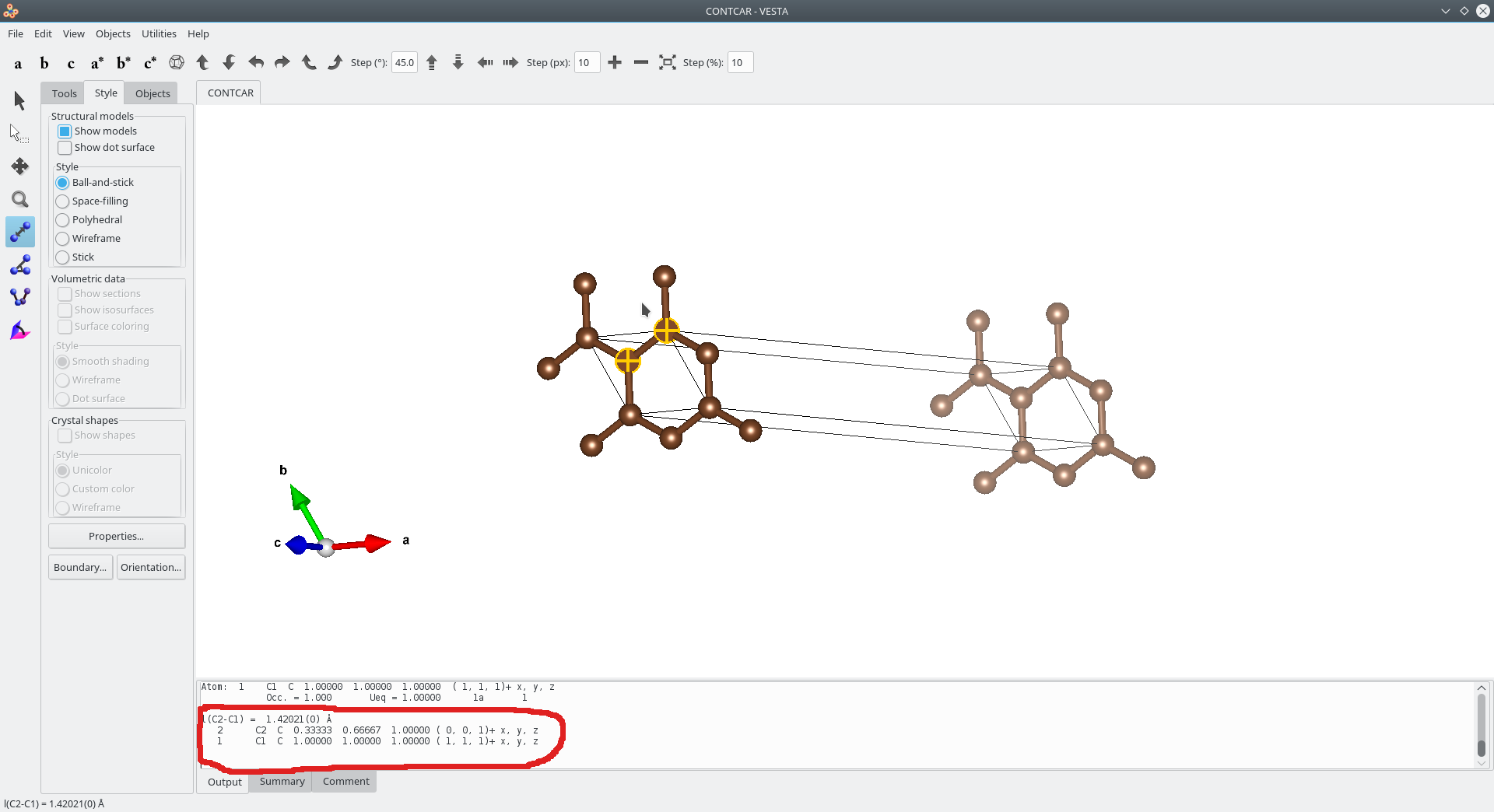
可以看出我们建立的模型理论上应该是正确的。
接下来是确定能带结构,首先将上面最开始的四个文件(INCAR、POSCAR、KPOINTS以及POTCAR)复制到另外一个文件夹,并且将生成的CHGCAR一起拷贝到另外一个文件夹。并将INCAR的内容改为如下所示
SYSTEM = Graphene molecule in a box !注释行
ISTART = 1 #随机初始化波函数
ICHARGE = 11 #从CHGCAR读取电荷分布
ENCUT = 300 #截断能
EDIFF = 1E-6 #能量收敛范围
ISMEAR = 0 ; SIGMA = 0.01
EDIFF = 1E-6将KPOINTS改为如下内容
K-POINTS !注释
10
line
reciprocal
0.0 0.0 0.0
0.0 0.5 0.0
-0.333333333333 0.6666666666667 0.0
0.0 0.0 0.0最后发现模拟出来的能带图不正确,就不贴图了,源码在附件可以下载,有懂得大佬可以自己研究。
接下来进入InSe的模拟,跟石墨烯类似,只需要改改相关参数,修改一些值就行了。其中相关的参数从这篇论文获得,首先是静态自洽处理得到电子的电荷,CHGCAR这个文件。
请注意:以下程序还未通过测试。
INCAR文件内容如下
SYSTEM = InSe molecule in a box !注释行
ISTART = 0 #随机初始化波函数
ICHARGE = 2
ENCUT = 300 #截断能,论文上写的是520ev,但是In和Si加起来最大306,用520时总是出错
EDIFF = 1E-5 #能量收敛范围
ISMEAR = 0 ; SIGMA = 0.01部分参考自这篇论文,并且这篇文章中有详细的模拟后的结果。由于本人技术有限,模拟出来的效果欠佳。可能是文件参数设置的有问题。
POSCAR内容如下
fcc:
4
1.000000 0.000000 0.000000
-0.50000 0.866025 0.000000
0.000000 0.000000 8.400000
1 1
Direct
0.000000 0.000000 0.000000
0.333333 0.666667 0.000000KPOINTS内容如下
K-POINTS #注释
0 #k点数目,0表示自动生成
G #k点产生方法,第一个字母有用
10 10 10
0 0 0将In和Se的POTCAR文件合成一个。直接将一个文件的内容复制到另一个末尾就行了, 要注意先后。
将得到的CHGCAR得到与前面四个文件一起复制到另一个文件夹。
最后面是源码,可能有错误。
如果在这个过程中遇到了其它问题,欢迎在评论区留言,或者Google一下,也欢迎把具体的解决方法留在评论区,以供后来者参考
欢迎转载,不需注明出处,就说是你写的
参考
附件下载:
感谢您的支持,请扫码打赏

